Illumina HiSeq/MiSeq 配对末端读长是什么样的?
What do Illumina HiSeq/MiSeq paired end reads look like?
我的理解是来自 Illumina HiSeq/MiSeq 平台的配对末端读取看起来像这样:
R1:
AAAAAACCCCCC
R2:
GGGGGGTTTTTT
R2 中发现的读数是 R1 中发现的读数的反向补充。然而,对于我的测序数据,情况似乎并非如此。如果有帮助,我从下面运行的一个 MiSeq 中得到一个读取对。
R1:
@M01814:86:000000000-A6MU9:1:1101:15397:1339 1:N:0:2
TACTCGCACCTATCCGGCACAGCAACACCATCTGGGGCTGAATCGCAATAGCATCTCTCACTTCCTCCATATCAGATTGCTCAAGGCAAGCACTACGCTGCAGTGCCCTCCACTCCCAATTCCCTGATGCTGGTCGTAACTTGCCACACCA
+
>>AA?BBBBBFFGGG2EEEGFBGHHHGA2FGHBGHF2EE?GHGHHFFEEHDGHEFGF5FEEFBGHGBCB5FHHH5F553@434FF31G11??233B1/1/?333B?3FB?/B24B2/2B2?44?3?23333B223<>@0CB22@2@F0/?/
R2:
@M01814:86:000000000-A6MU9:1:1101:15397:1339 2:N:0:2
TAAGGGGCCTAGAACAGGCACCATACATTCAATTGGCTGTGGCAAGTAACAACCAGCATCAGGGAATGTGGAGTGGAGGGCACTGCAGCGAATTGCTTGCCTTGAACAATCTTATATGGGGGAAGTAGACGAACCAATGTGGAGTCAGCCC
+
>AA>>>ADDAFFGGGGG4FGGGFHFHFHHHFHHHB3B32EFBGGE25FGHHHHACEGG533BAGFFF355331BG1@1>EF1E23F333/>//134B43?F34B3334B334444?443B?/<C/23333////<0/<11111/?01?G0?
简短回答:通常 R1 和 R2 不是彼此的反向互补。
更长的答案:
reverse reads是以反向方式排序的,但是reverse reads的内容不一定是forward reads的反向补码。
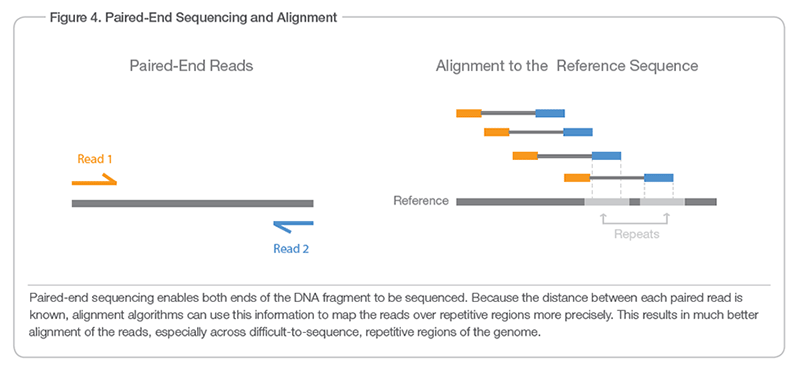
大多数情况下,您想要测序的 DNA 片段比 MiSeq 实际可以测序的 ~100bp(或最多 300bp,取决于来源)长很多。因此片段的末端被排序,你只知道正向和反向读取的序列以及它们相距多远(如果我没记错的话,内部配对距离)。 This graphic Illumina 网站显示。
假设您可以对 10bp 进行测序,并且想要对长度为 25 的片段进行测序:
---r1---->
AAAAACCCCCGGGGGTTTTTAAAAA
<----r2---
在这种情况下,您的内部配对距离为 5(读取之间未排序的碱基的 nr),您将无法获得有关读取之间序列的信息(在本例中为所有 G)。如果您像这样分析较小的片段大小
---r1---->
AAAAACCCCCGGGGG
<----r2---
你的读数重叠,你得到一个负的内部配对距离。然后你会得到一些你描述的冗余信息,但通常情况并非如此。
您可以找到另一篇关于方式的有用文章 here。
希望对您有所帮助。
我的理解是来自 Illumina HiSeq/MiSeq 平台的配对末端读取看起来像这样:
R1:
AAAAAACCCCCC
R2:
GGGGGGTTTTTT
R2 中发现的读数是 R1 中发现的读数的反向补充。然而,对于我的测序数据,情况似乎并非如此。如果有帮助,我从下面运行的一个 MiSeq 中得到一个读取对。
R1:
@M01814:86:000000000-A6MU9:1:1101:15397:1339 1:N:0:2
TACTCGCACCTATCCGGCACAGCAACACCATCTGGGGCTGAATCGCAATAGCATCTCTCACTTCCTCCATATCAGATTGCTCAAGGCAAGCACTACGCTGCAGTGCCCTCCACTCCCAATTCCCTGATGCTGGTCGTAACTTGCCACACCA
+
>>AA?BBBBBFFGGG2EEEGFBGHHHGA2FGHBGHF2EE?GHGHHFFEEHDGHEFGF5FEEFBGHGBCB5FHHH5F553@434FF31G11??233B1/1/?333B?3FB?/B24B2/2B2?44?3?23333B223<>@0CB22@2@F0/?/
R2:
@M01814:86:000000000-A6MU9:1:1101:15397:1339 2:N:0:2
TAAGGGGCCTAGAACAGGCACCATACATTCAATTGGCTGTGGCAAGTAACAACCAGCATCAGGGAATGTGGAGTGGAGGGCACTGCAGCGAATTGCTTGCCTTGAACAATCTTATATGGGGGAAGTAGACGAACCAATGTGGAGTCAGCCC
+
>AA>>>ADDAFFGGGGG4FGGGFHFHFHHHFHHHB3B32EFBGGE25FGHHHHACEGG533BAGFFF355331BG1@1>EF1E23F333/>//134B43?F34B3334B334444?443B?/<C/23333////<0/<11111/?01?G0?
简短回答:通常 R1 和 R2 不是彼此的反向互补。
更长的答案: reverse reads是以反向方式排序的,但是reverse reads的内容不一定是forward reads的反向补码。 大多数情况下,您想要测序的 DNA 片段比 MiSeq 实际可以测序的 ~100bp(或最多 300bp,取决于来源)长很多。因此片段的末端被排序,你只知道正向和反向读取的序列以及它们相距多远(如果我没记错的话,内部配对距离)。 This graphic Illumina 网站显示。
{kind=link}
假设您可以对 10bp 进行测序,并且想要对长度为 25 的片段进行测序:
---r1---->
AAAAACCCCCGGGGGTTTTTAAAAA
<----r2---
在这种情况下,您的内部配对距离为 5(读取之间未排序的碱基的 nr),您将无法获得有关读取之间序列的信息(在本例中为所有 G)。如果您像这样分析较小的片段大小
---r1---->
AAAAACCCCCGGGGG
<----r2---
你的读数重叠,你得到一个负的内部配对距离。然后你会得到一些你描述的冗余信息,但通常情况并非如此。
您可以找到另一篇关于方式的有用文章 here。
希望对您有所帮助。